It seems @jamesbradbury is on fire. Early examples moved here from another thread to stay focus 
Thanks PA! Copied here.
CLI is great.
I made two bash scripts that you can add to your .bash_profile . These do batch processing using nmf or hpss functions.
The two functions would then be called through your terminal of choice (assuming you are using bash and not zsh or fish or something) as batch_nmf and batch_hpss .
batch_nmf takes three arguments.
1: Input Folder
2: Output Folder
3: Number of Ranks
The output of dog.wav would be dog-nmf.wav in your output folder.
batch_hpss takes two arguments
1: Input Folder
2: Output Folder
The output of cat.wav would be cat-h.wav and cat-p.wav in your output folder.
Code below. Enjoy!
### batch_hpss is passed two variables.
### Arg 1: Source Folder
### Arg 2: Output Folder
function batch_hpss () {
shopt -s nullglob
if [ -d "$2" ]; then
for src in $1/*;
do
file_name=$(basename "$src")
strip_ext=$(echo "$file_name" | cut -f 1 -d '.')
harm_sf="$2/$strip_ext-h.wav"
perc_sf="$2/$strip_ext-p.wav"
echo "Starting separation on $src"
hpss -source $src -harmonic $harm_sf -percussive $perc_sf
echo "$src has been separated!"
echo "Output at $2_filename_p/h.wav"
done
echo "Finished batch hpss processing"
else
echo "The output directory does not exist."
echo "I have made it for you!"
echo "You will need to re-run the command in your shell."
mkdir $2
fi
}
### batch_nmf is passed three variables.
### Arg 1: Source Folder
### Arg 2: Output Folder
### Arg 3: Number of NMF Ranks
function batch_nmf () {
shopt -s nullglob
if [ -d "$2" ]; then
for src in $1/*;
do
file_name=$(basename "$src")
strip_ext=$(echo "$file_name" | cut -f 1 -d '.') ## Remove extension
out="$2/$strip_ext-nmf.wav" ## Make name for output
## Process ##
echo "Starting NMF on $src"
nmf -source $src -rank $3 -resynth $out
echo "Completed NMF on $src"
echo "Output at $out"
done
echo "Finished batch nmf processing"
else
echo "The output directory does not exist."
echo "I have made it for you!"
echo "You will need to re-run the commannd in your shell."
mkdir $2
fi
}
To clarify, you need to copy this code to your clipboard. Open up terminal go to your root directory and type open .bash_profile. If the file doesn’t exist type touch .bash_profile, then repeat the previous command. Once you’ve opened the bash_profile paste the contents of the clipboard into that file, save and close it. Run the command . .bash_profile or close and restart terminal. The commands should now work!
1 Like
Okay one more for fun.
This is a server in python that can dispatch asynchronous calls to your shell. A folder is watched for any files that are added to it and if it detects a new file it automatically calls the shell to run hpss on it. You can decide if you want the output to be relative to the file or in an absolute location. The best part is, you don’t have to wait for one process to finish before you add another file - you can just keep adding files to the folder and it will queue it up. The next stage is to make it run multiple processes simultaneously 
This one isn’t super friendly if you haven’t used python before and I definitely haven’t tested it. It will try and hpss anything you put in the file, and there is no protections against this however the script probably wont execute which won’t stop the overall server from running.
This requires Python 3.6 and watchdog which can be installed with pip install watchdog
import logging
import sys
import time
import os
import subprocess
from watchdog.events import FileSystemEventHandler
from watchdog.observers import Observer
logging.basicConfig(level=logging.DEBUG)
class MyEventHandler(FileSystemEventHandler):
def __init__(self):
self.creation_rule = ''
self.file_name = ''
self.only_file = ''
self.dir_name = ''
self.base = ''
self.abs_dir = None
def catch_all_handler(self, event):
logging.debug(event)
def on_moved(self, event):
self.catch_all_handler(event)
def on_created(self, event):
self.catch_all_handler(event)
self.file_name = str(event.src_path)
self.base = os.path.basename(self.file_name)
self.only_file = os.path.splitext(self.base)[0]
if self.creation_rule == 'Relative':
self.dir_name = f'{os.path.dirname(self.file_name)}/{self.only_file}_proc'
try:
os.makedirs(self.dir_name)
except FileExistsError:
print('Files possibly being overwritten or already exist')
pass
elif self.creation_rule == 'Absolute':
self.dir_name = f'{self.abs_dir}/{self.only_file}_proc/'
if self.abs_dir != None:
try:
os.makedirs(self.dir_name)
except FileExistsError:
print('Files possibly being overwritten or already exist')
pass
cmd = ['hpss', '-source', self.file_name, '-harmonic', f'{self.dir_name}/{self.only_file}-harm.wav', '-percussive', f'{self.dir_name}/{self.only_file}-perc.wav']
subprocess.call(cmd)
def on_deleted(self, event):
self.catch_all_handler(event)
def on_modified(self, event):
self.catch_all_handler(event)
### Set your path here that you want to watch. Make sure you leave the backslash off at the end
path = '/Users/jamesbradbury/dev'
event_handler = MyEventHandler()
event_handler.creation_rule = 'Absolute' ### You can set this string to 'Relative' or 'Absolute' to change how the output file structure works
event_handler.abs_dir = '/Users/jamesbradbury/_abs_processing'
observer = Observer()
observer.schedule(event_handler, path, recursive=False)
observer.start()
try:
while True:
time.sleep(1)
except KeyboardInterrupt:
observer.stop()
observer.join()
2 Likes
oh la la, you’re on fire! I look forward to batch process some sounds now
Quick note: the arguments in the CLI are in line, spelling included, with the Max ones. (all lower case, not hard to guess from the SC helpfile) and the right format (for -fftsettings you need the 3 windowing arguments as one)
Just trying it, but getting syntax error messages. What is the right way of doing it?
I tried the paths variables with no quotes, single quotes and double quotes, but none of them is accepted
batch_nmf("/Users/hans/Desktop/flucoma-input" “/Users/hans/Desktop/flucoma-output” 5)
-bash: syntax error near unexpected token `"/Users/hans/Desktop/flucoma-input"’
I didn’t get a notification about this Hans, so I’m sorry for the delayed response.
The way that bash takes arguments is positional and with no brackets.
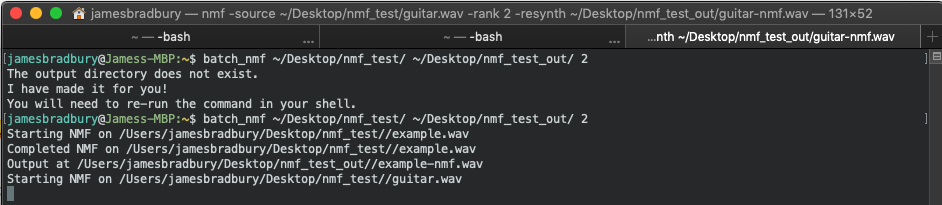
You can see in this example I have tried to run the batch process but the output folder isn’t made yet. Bash makes it for you with mkdir and then you can run the process again.
I’ve also uploaded a short video to clarify usage
If this is useful for you, I’ll add some more functionality to pass arguments in an easy way. Just let me know.
1 Like
ok, that helpful
I did the following steps from your earlier message
hans$ open .bash_profile
I copied your code in there, saved, quit terminal, reopened.
But I’m still getting an error message:
MacBook-2018:~ hans$ pwd
/Users/hans
MacBook-2018:~ hans$ batch_nmf ~/Desktop/flucoma-input ~/Desktop/flucoma-output 5
Starting NMF on /Users/hans/Desktop/flucoma-input/Oud-normal-Csharp3-ff.wav
-bash: nmf: command not found
Completed NMF on /Users/hans/Desktop/flucoma-input/Oud-normal-Csharp3-ff.wav
Output at /Users/hans/Desktop/flucoma-output/Oud-normal-Csharp3-ff-nmf.wav
Finished batch nmf processing
MacBook-2018:~ hans$
Can you post the contents of your .bash_profile here? I have a feeling I know what the problem is, but can’t confirm till I see. Another thing to check is can you do basic commands like ls and cd. If you can’t there is something wrong with your $PATH.
Okay, so the bash_profile is configured correctly that isn’t the issue. The problem is that you don’t have the flucoma stuff in your path somewhere and so when the bash script tells the shell to run nmf, it has no idea what you’re talking about.
Easiest way to add it to your shell is to put it somewhere you know it won’t move. Mine is in a folder close to my ~/ directory.
run sudo nano /etc/paths in your terminal, put in your password. A text editor will come up. Add a line at the bottom with the directory of the flucoma tools.
It should look something like this:
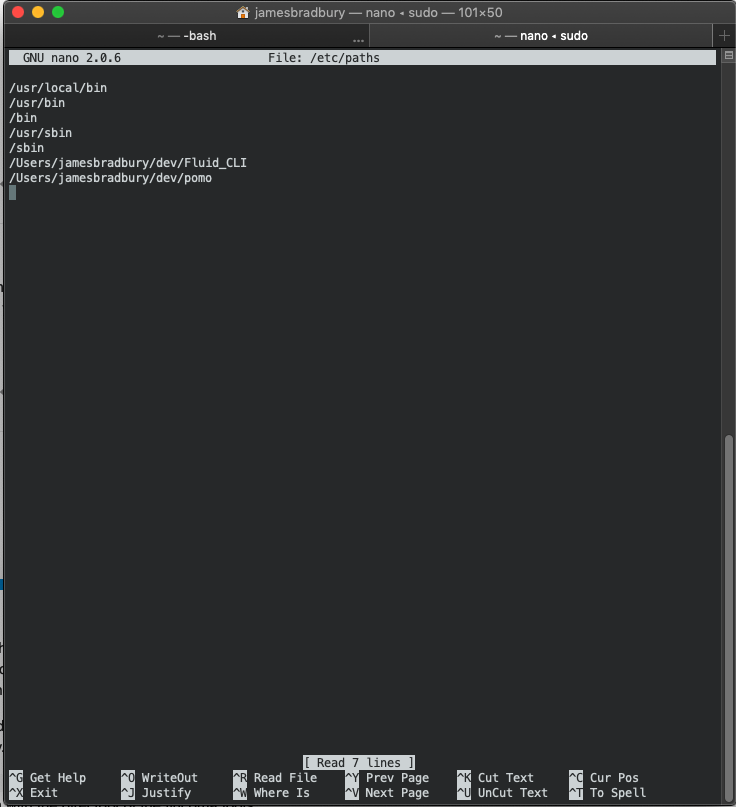
You can see I have the CLI stuff in a folder called Fluid_CLI.
Once you have put that directory in, Control + O then enter then restart your terminal. You then should be able to use the flucoma cli.
Genial! Thanks a lot for your trouble-shooting tips.
I will assume this means its working for you
Thanks for the film @jamesbradbury!
The original bash scripts I made were not very extensible. In particular, adding -parameters was proving very hacky and not very fluent. Every Mac (sorry windows users) comes with python 2 so I’ve made some scripts with this that can be used for batching some of the processes. At the moment this includes only nmf, but from the plenary there seems to be a lot of interest in this particular tool.
Usage is simple, put the scripts next to the executables and run them from them from your terminal.
Example file structure:
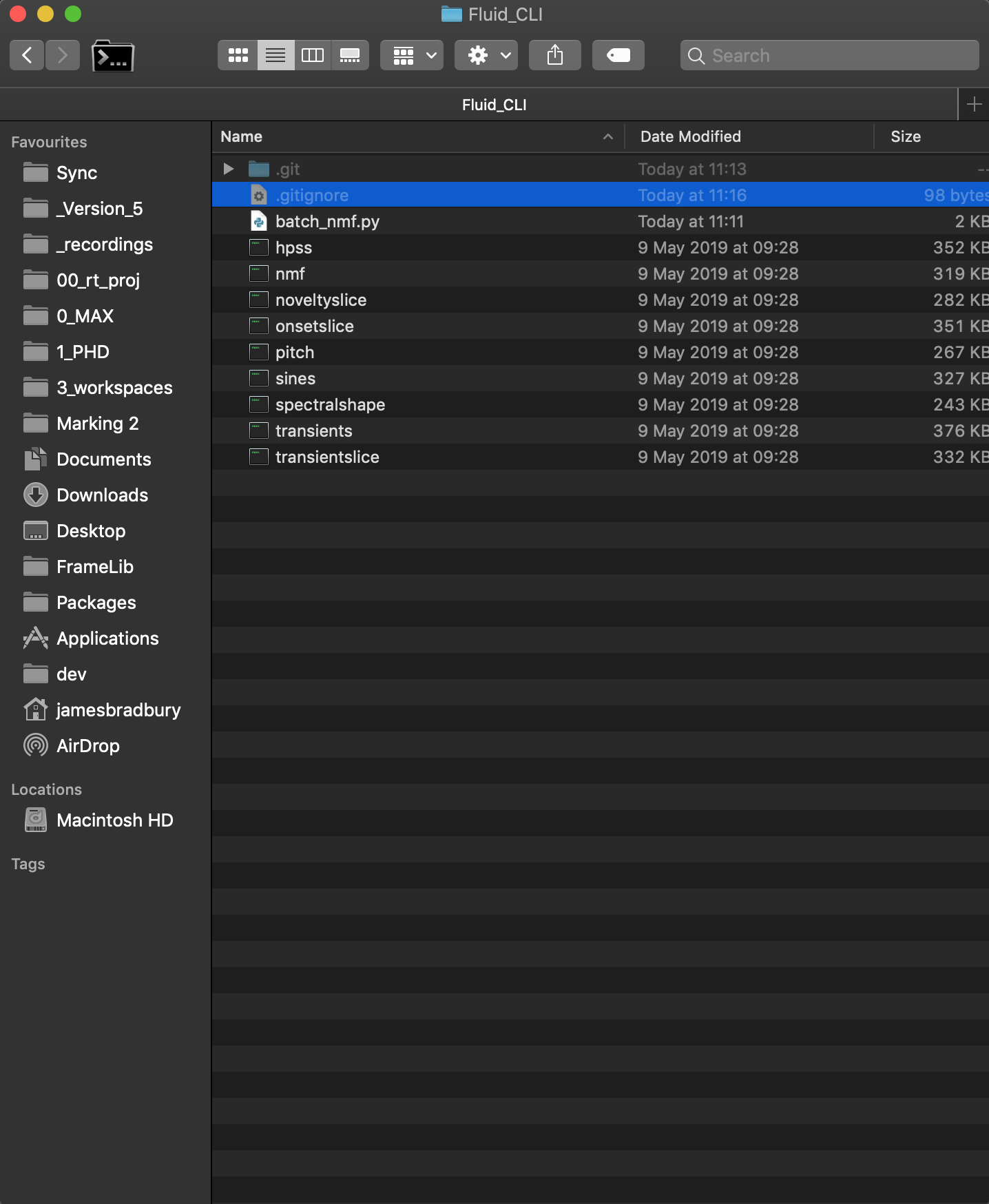
Really, the scripts can be anywhere. You just need to remember where you put them!
Example usage:
First, you have to cd to the location of the scripts. I put mine next to the executables so lets go there in the terminal.
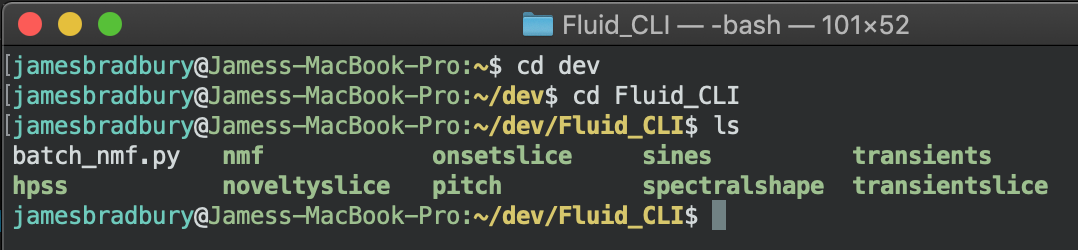
Then we call the script with some parameters. Note, the input and output parameters are not the same as the original ones. We are dealing with input and output folders and the parameters reflect this as -input and -output.
Link to the scripts.
Any questions/suggestions, shoot!
2 Likes